Raw sequencing data from Wang et al 2017 were downloaded and re-mapped to HuSjV2 genome. Normalised expression based on library-size and gene length were calculated using edgeR. MDS and PCA representations were added.
In the original paper, the data were represented as de novo assemblied transcripts using Trinity. There were in total 48 RNA-seq samples including male and female Sj worms at different timepoints (dpi): 14, 16, 18, 20, 22, 24, 26, 28, each with 3 biological replicates.
I downloaded all SE fastq files and re-mapped to the HuSjV2 genome using STAR, count reads using featureCount, and processed the raw counts using edgeR to obtain normalised expression (on library size and gene length). The processed data can be compared across samples and genes.
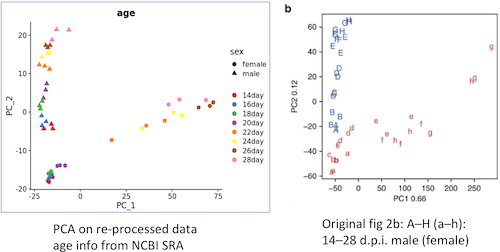
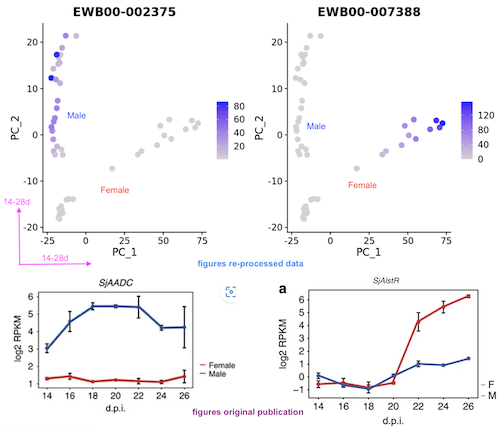
Based on the metadata from NCBI SRA, the re-processed data agrees well with the original paper, in terms of pca representation. I also checked two representative genes in the paper, SjAADC and SjAlstR, the expression also fits well.
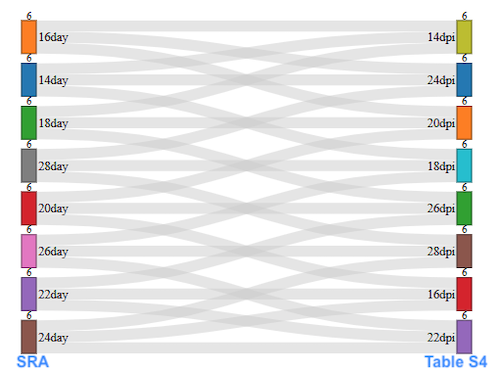
However, there seems a mistake in Supplementary Table 4 for sample age information. The table doesn’t agree with NCBI SRA and pca different from Fig. 2b. Just a reminder to use information from SRA.